Tutorial
Search
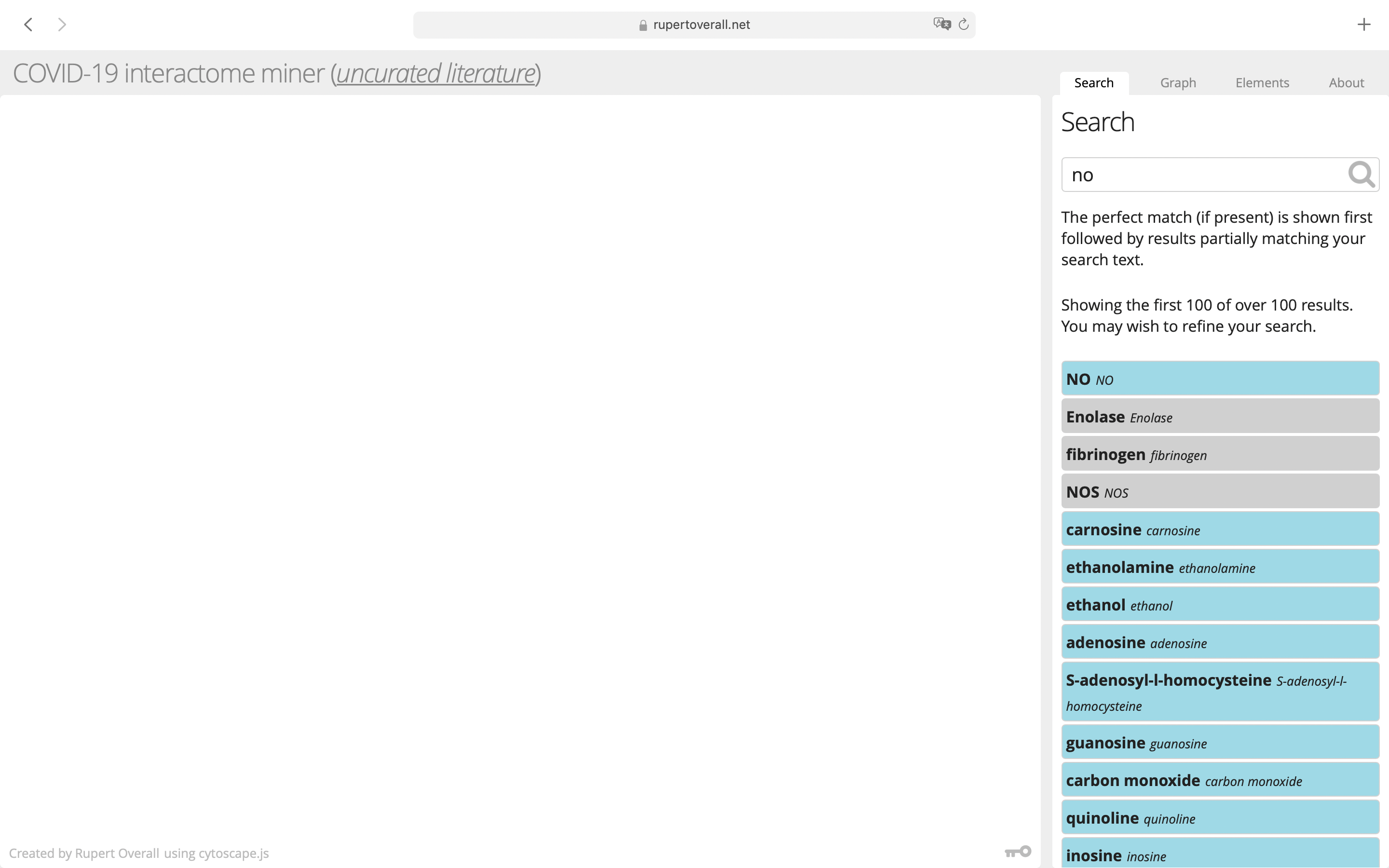
Start in the Search tab and type the name of a gene/protein or biological process of interest into the search box. Click on the search icon or hit enter to confirm. The database will be searched for your term and, if found, the corresponding entry will be returned, together with all of the entities that are connected to it, as a graphically-displayed network. If your search term matches several entries in the database, they will be all returned in a list and you can select the one that best corresponds to your intended search. If there is no match in the database, then nothing is returned. Not all proteins etc. have been mentioned in the COVID-19 literature, so many of your favourite genes will not be found.
Navigate the network
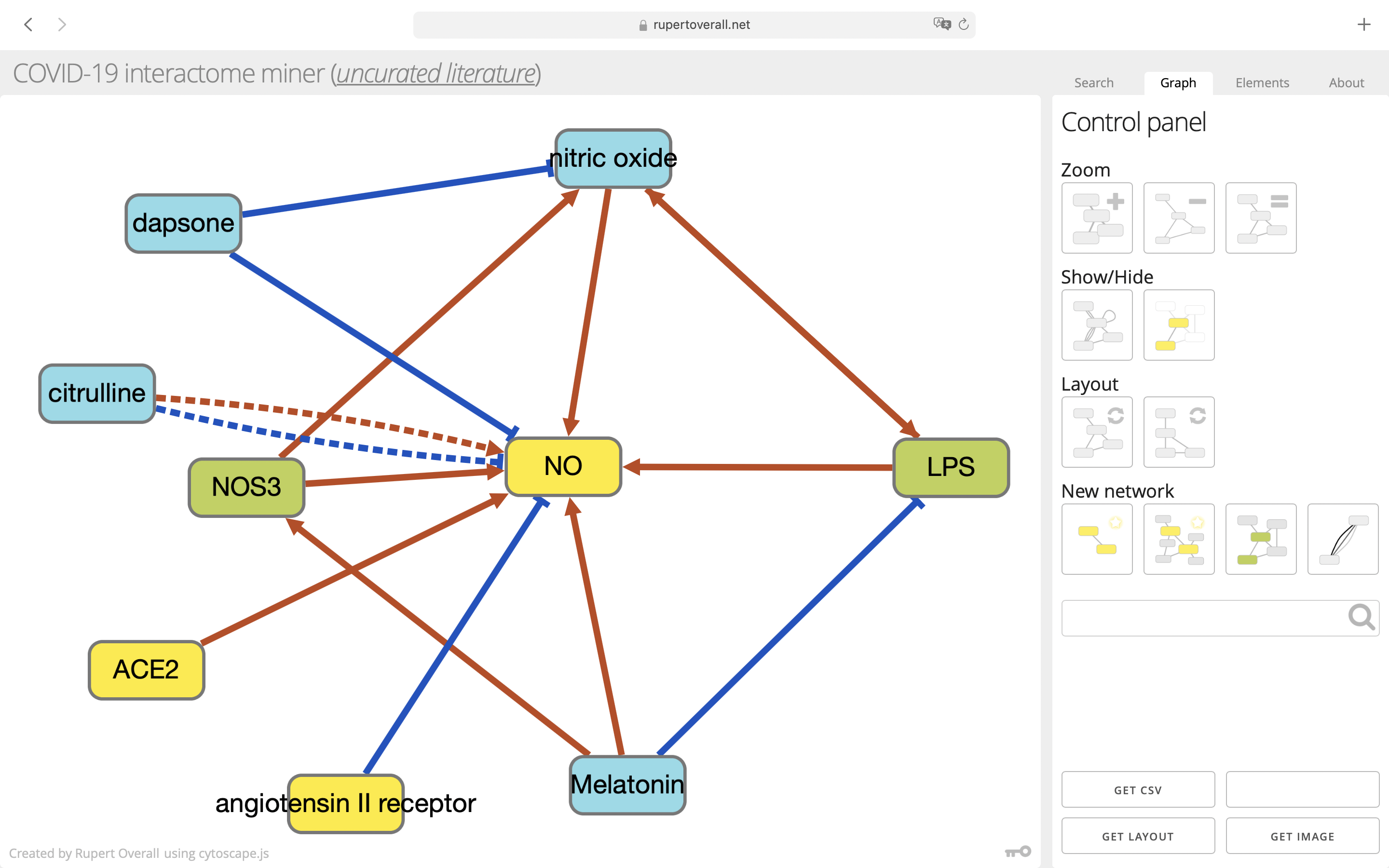
Once you have loaded a network, you will be automatically switched to the Graph tab where there are tools for navigating the network graph. For some queries, there may be very many entities (nodes) and it can be confusing to see what's going on—the tools described below should help you sort through the data.
Zoom



Using the buttons in the Zoom group, you can increase or reduce node size so that the nodes and their labels don't overlap so much and become more readable. Click zoom in to increase or zoom out to decrease the node size. You can always click reset zoom to go back to the default size. Note that this does not actually properly zoom the network—that is done by scrolling with your mouse or trackpad.
Show/Hide


Replication of connections occurs when more than one manuscript provides evidence of the same interaction—so this can be a hint that the interaction might be real and/or important (remember this is automated text mining and includes much non-peer-reviewed literature). By default, multiple interactions between the same nodes have been collapsed so that the width of the connection reflects how many replicate connections there are. In order to follow up the links to individual literature sources, you will need to 'expand' the connections using the expand button in the Show/Hide group so that all of the replicate interactions are shown—they will all have different literature links. When you activate the expand button, it changes into the collapse button. Clicking this collapses the multiple interactions back into composite connections.
Note: it is also possible to quickly expand a single composite connection simply by clicking on it. Once it is expanded, further clicks on the individual connections will switch to the Elements tab and activate the Connections browser (see below)
To better focus on a particular interaction, or subset of interactions, you can click on several nodes while holding down the <shift> key. This will select (highlight) the nodes. The hide unselected nodes button (with the yellow nodes) will now hide the non-selected nodes so you can focus on just the ones you're interested in. You can re-run the layout to fit this new sub-network to the screen area (see below) and perform all other operations. When there are hidden nodes, the button becomes the show all nodes button and clicking on this will reveal the rest of the network again.
Layout


The buttons in the Layout group will re-run the layout algorithm to place the nodes in a particular way and fit the network to the available space on the screen. The default 'force-directed' layout will push the nodes apart to make them as far apart as possible (and involves a random step so the layout will be different each time you run it), whereas the 'grid' layout places nodes on a regular grid, which can be neater, but makes the relationship between nodes a bit less obvious.
New network




These functions reload the page with a new network. Selected nodes can be used to make a simplified sub-network containing only the selected nodes, or to make an extended sub-network that contains the selected nodes and their neighbour nodes. Be aware, this latter option can yield very large networks, so it is not advised to use this with more than one or two selected nodes.
Note: very large networks (with thousands of connections) may load very slowly on your computer—the drawing of the embedded network is quite complex. If more than 10000 interactions are retrieved from the database, then you will receive an error message. Most browsers will not handle a webpage this complex.
By default, unknown (‘unmapped’) nodes are not shown but this can be changed using the show unmapped nodes button. Clicking on this button will reload the page to show a new network including all nodes (both mapped and unmapped) and the button will now allow you to hide unmapped nodes. For information about what ‘unmapped’ nodes are, see the note about mapping below.
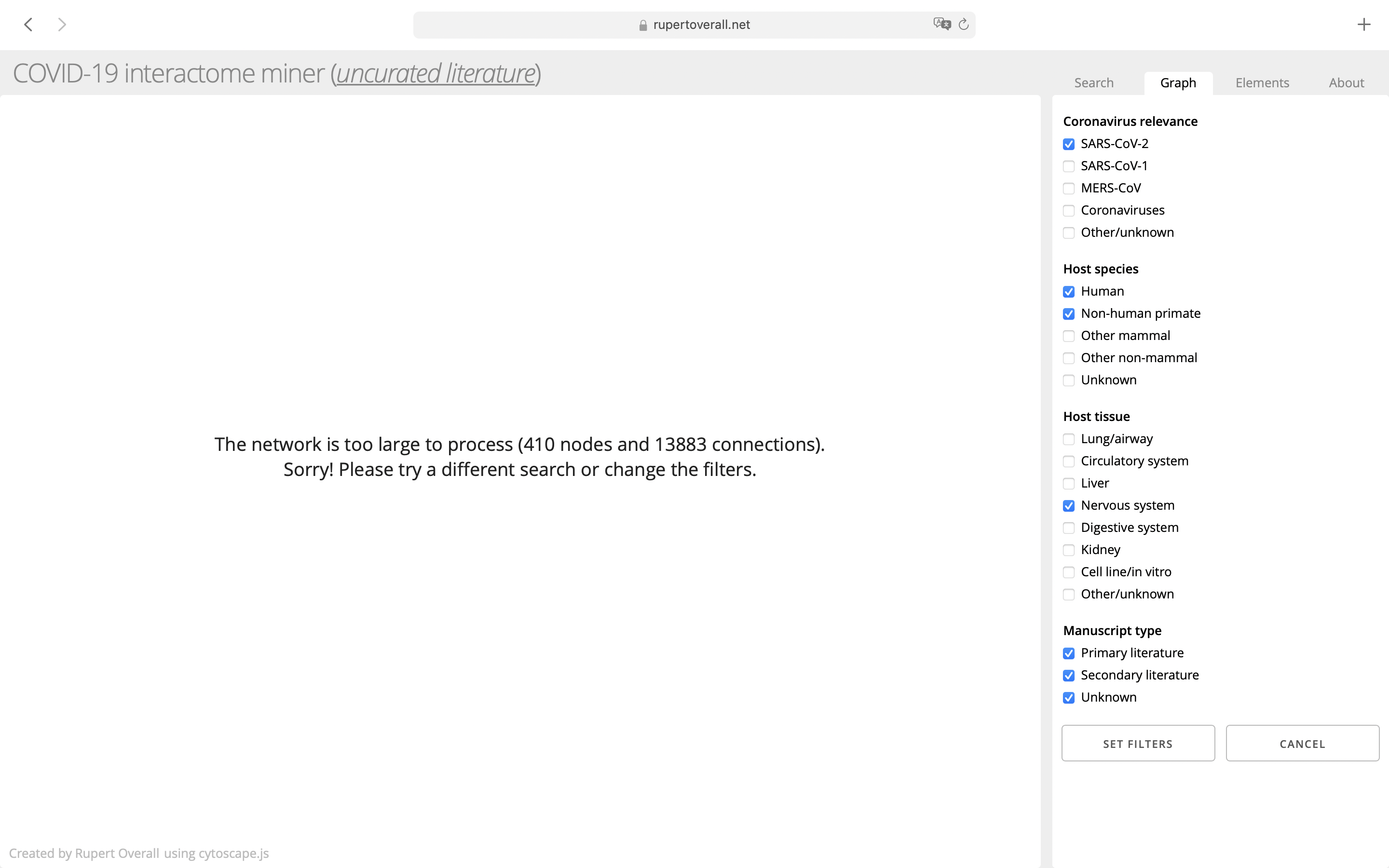
Clicking on the tag filter button will bring up a panel allowing a filter for each tag to be switched on or off. Clicking ‘Set Filters’ will update the filters so that only the connections with the activated tick boxes are shown. Clicking ‘Cancel’ will dismiss this panel without changing the filter settings. These filters will persist for the current browser session.
Note: At least one checkbox must be activated for each tag. Otherwise you set all connections to be hidden and you will see nothing!
To learn about tags, see the section Tags below.
Network search

There is a text entry field in the graph control panel which allows quick searching of the visible network. Sometimes the networks can be large and it is hard to find nodes of interest. If you know the name, just start typing in the search box and a list of matching names will appear. Only the visible nodes in the currently loaded network are searchable, so you know if the name appears in that list, the node is in the network somewhere. When you find the name of the element you want, select it from the list and the corresponding node will be selected. You can do this for as many nodes as you like and they will all be selected one after the other.
Export options



It is possible to download a network in several different formats. Note that the downloaded network will only include nodes that are currently visible on the network canvas. The data can be saved as a CSV file (comma-delimited text file that can also be opened in Excel) containing a table all of the interactions, as a JSON file describing the underlying Cytoscape.js graph, or as a screenshot of the current graph window (in PNG format).
Elements
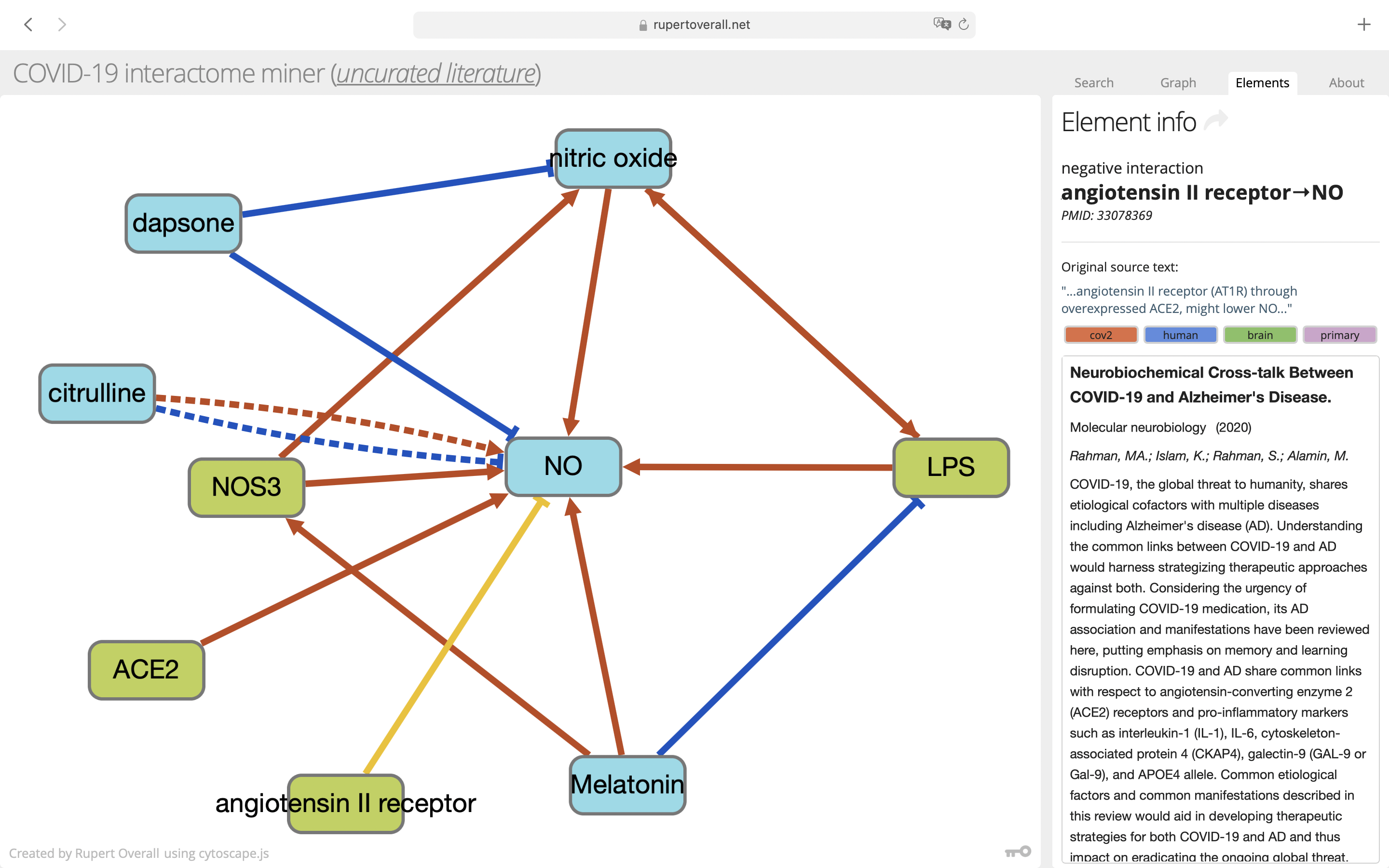
The elements tab is used to show detailed information about the currently-selected graph element (a node or a connection). If you click on a node or a connection, you will be automatically switched to this tab (you can always switch manually between tabs at any time).
Nodes
For a node, the node name and class (whether it is a gene/protein, a chemical, a biological process, or some other unknown type) is shown as well as its accession code in the most appropriate database. The entry in the relevant database is loaded and shown in the information field below. To load this page in a new browser tab, click the 'linkout' arrow next to the Element info text or you can also click on the link identifier. For some entities, there is no mapping to an external database, so this information is not available. Please see the notes about mapping below.
Connections
For a connection, the information shown includes the source and target nodes and whether it is a positive or negative interaction. Positive interactions (red lines) are shown as arrows in the graph, whereas lines representing negative interactions (in blue) end with a 'T'. Interactions depicted with broken lines are derived from non-peer-reviewed preprints. Please note that for collapsed connections (see Show/Hide above), both arrow endings may be shown; this happens because the arrows are simply drawn on top of each other and different literature sources can propose different interactions between the same entities.
The details of the interaction have been extracted from the source literature by automated text mining. The fragment of text used by the text mining software is shown in the 'original source text' field. You can usually immediately tell from this text fragment whether the tex mining software has understood the text correctly. You can use this information (usually without needing to dig into the source manuscript) to decide whether the interaction is suitable for your curation effort. To load this page in a new browser tab, click the 'linkout' arrow next to the Element info text or you can also click on the link identifier.
Tags
Each interaction is associated with tags generated from its source manuscript. These tags quickly reveal some key information about the source manuscript and are displayed in the Element information panel when a connection is selected (see the section Connections above). The tags can also be used to filter the connections and hide those that you might not be interested in. For details on using the tag filter, see the instructions above. There are four classes of tag;
- Relevance: This refers to how relevant the manuscript might be to SARS-CoV-2. The COVIDminer database contains a large corpus of literature covering many different viruses and coronavirus-related fields. This can be useful in discovering links between entities which have not yet been established for the SARS-CoV-2 itself.
- Species: Much research, especially on related viruses, has been done in species other than human. Many pathways in other organisms are likely also to be relevant to humans, so a broad search might identify novel interactions.
- Tissue: Some of the most important tissues in SARS-CoV-2 infection have been tagged to help narrow down your search.
- Manuscript type: Where possible, a distinction has been made between primary, experimental, literature and secondary literature (reviews, perspective/commentary pieces etc.).
Key
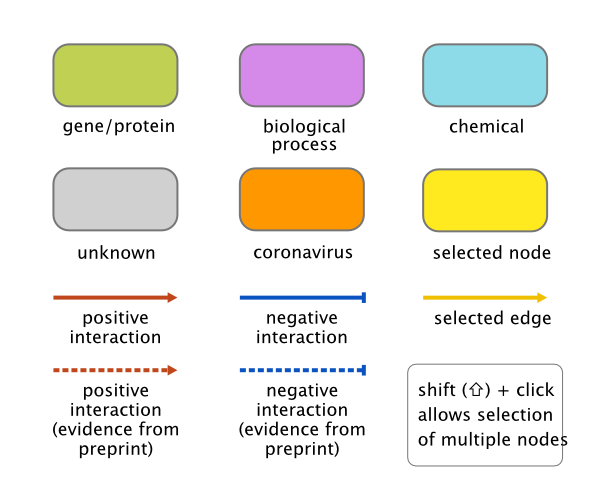
Each of the colours used for the components in the networks carries some meaning. The different element types are described here:
- Gene/protein: All mentions of genes or gene products (RNA, proteins) are mapped to the corresponding gene identifier. In addition, genes and proteins from different organisms (or where the organism is unclear) are mapped to the homologous human gene. This means that potential interactions discovered in closely-related species will be visible when searching for interaction partners of human protein identifiers.
- Biological process: This class includes all process that have been mapped to an identifier such as the Gene Ontology and the NCBI Medical Subject Headings (MeSH).
- Chemical: Chemicals, metabolites and small molecules.
- Virus: Similarly to the situation with other genes/proteins, all viral genes/proteins have been mapped to SARS-CoV-2 identifiers. As this is a resource designed to find information relating to SARS-CoV-2, it makes sense to link viral gene products that may share similar function to a common node. This enables the discovery of new potential interactions in a research environment that is still very incomplete. Because there is currently no uniform mapping scheme available for SARS-CoV-2 and the commonly used gene/protein names are very ambiguous, there is still quite some difficulty in correctly identifying SARS-COV-2 information during text mining. Hopefully, the tools provided in COVIDminer make it easy to manually sort out the useful information. We are actively working on improving the mapping resources for SARS-CoV-2 and updated interaction data will be continuously added.
- Unknown: Finally, there are many concepts (particularly biological processes) for which there are no good mappings; i.e. it has not been possible to automatically assign the text to a biological entity. Where the natural language processing software has detected an entity that cannot be mapped, it will be presented in the network as plain text. Although there is no detailed metadata for such entities, they can often be informative enough to warrant a closer look at the source manuscript. It is common to see many separate entities with similar names where they should really be collapsed into one entity. By default, these nodes are hidden to reduce clutter in the network, but they may reveal useful links to novel concepts, so it is possible to show these using the show unmapped button described above. We are working to make these text-only elements more relevant and more informative.
A visual key can be called up at any time by clicking on the key icon in the bottom right corner of the network canvas.